

Kostas, convive con la beta talassemia major in Germania
Scopri di più sulle trasfusioni di sangue per la gestione della beta-talassemia qui.
Esistono diverse forme di beta-talassemia:1
- beta-talassemia non trasfusione-dipendente (anche detta “beta-talassemia intermedia”): le persone con questa forma di talassemia di solito presentano una lieve anemia;1 esse, talvolta, non necessitano di trattamento fino all’età adulta, sebbene il bisogno di trasfusioni di sangue possa aumentare con il passare degli anni;3,4
- beta-talassemia trasfusione-dipendente (anche detta “beta-talassemia major”): le persone con questa forma di beta-talassemia solitamente hanno un basso numero di globuli rossi a partire dai primi anni di vita.1,3 Forse avrai sentito il team medico utilizzare il termine “anemia”. I globuli rossi trasportano l’ossigeno in tutto il corpo, quindi, averne un numero ridotto può causare affaticamento (stanchezza) e affanno.1,4 Le persone con questo tipo di beta-talassemia necessitano di trasfusioni di sangue regolari per tutta la vita;3,4
- Portatori di beta-talassemia (“beta-talassemia minor” o “tratto talassemico”): una persona portatrice di beta-talassemia presenta, talvolta, una forma lieve di anemia, che raramente richiede un trattamento.2,3
Cosa causa la beta-talassemia?

Le persone con beta-talassemia presentano un numero ridotto di globuli rossi, ma qual è il motivo? Tutto inizia con un problema nella produzione di emoglobina.1
L’emoglobina è una proteina presente nei globuli rossi che trasporta l’ossigeno dai polmoni al resto del corpo.5 Ogni persona ha diversi tipi di emoglobina nel sangue, in proporzioni che dipendono dall’età.3,5 In condizioni fisiologiche, dopo la nascita i livelli di Hb totali sono mantenuti dall'emoglobina adulta (HbA).5
Normalmente, l’HbA è costituita da quattro catene proteiche globulari: due molecole di alfa-globina e due molecole di beta-globina.5 Nelle persone beta-talassemiche il rapporto 1:1 di alfa-globina e beta-globina è sbilanciato.1
Vediamo come può succedere.
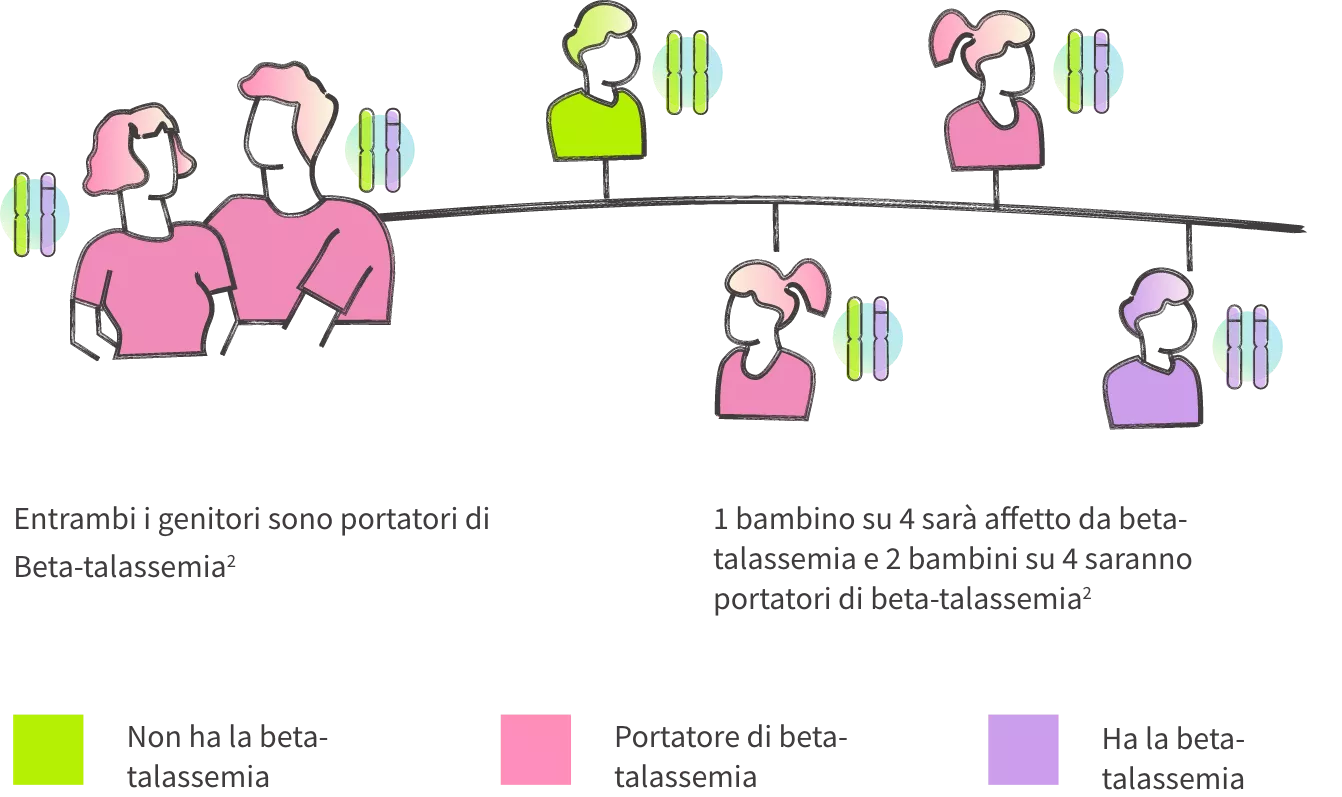
- Ogni individuo eredita i propri geni per metà dalla madre e per metà dal padre.6 I geni sono frammenti di DNA che indicano all’organismo come produrre una specifica proteina.6 Le proteine sono sostanze presenti in tutti gli organismi viventi, che svolgono funzioni essenziali per la vita.7
- Le persone con beta-talassemia trasfusione-dipendente o non trasfusione-dipendente presentano normalmente due geni per la beta-globina (HBB) mutati o “difettosi”, uno da ciascun genitore.1,3 Le variazioni all’interno dei geni HBB fanno sì che l’organismo produca una quantità inferiore di proteina beta-globina o non ne produca affatto.1,2
- Senza una quantità sufficiente di beta-globina, si possono verificare due condizioni:
- Con i globuli rossi che muoiono precocemente e una quantità insufficiente di emoglobina, l’organismo può non avere abbastanza globuli rossi per fornire ossigeno in modo efficace a tutte le sue parti.1,5
I portatori di beta-talassemia hanno un gene HBB sano e uno modificato.2

Quali sono i sintomi della beta-talassemia?
I sintomi della beta-talassemia dipendono dalla forma di malattia: le persone affette possono manifestare sintomi da moderati a gravi.1
Sintomi della beta-talassemia trasfusione-dipendente (beta-talassemia major)1
- Pallore
- Debolezza muscolare
- Rallentamento della crescita
- La pelle o il bianco degli occhi diventano gialli (ittero)
- Ulcere alle gambe, ovvero ferite aperte, spesso dolorose, sulla pelle
- Deformazioni ossee
- Milza più grande del normale
- Problemi al fegato
- Complicanze da sovraccarico secondario di ferro dovute alla trasfusione
Senza trasfusioni regolari, è possibile avvertire anche stanchezza (affaticamento)4 o sviluppare ematopoiesi extramidollare (quando i globuli rossi si formano al di fuori del midollo osseo).9
Sintomi della beta-talassemia non trasfusione-dipendente (beta-talassemia intermedia)1
- Complicanze da sovraccarico di ferro
- Ulcere alle gambe, ovvero ferite aperte, spesso dolorose, sulla pelle
- Deformazioni ossee
- Osteoporosi, una condizione in cui le ossa si indeboliscono e hanno maggiori probabilità di rompersi
- Compressione del midollo spinale
- Milza più grande del normale
- Calcoli biliari, ovvero piccoli “sassolini” nella cistifellea (un piccolo organo vicino al fegato)
- Trombosi, ovvero un coagulo di sangue all'interno di un vaso, che ostacola la circolazione

Naziha, convive con la beta talassemia major in Francia
Se ricevi trasfusioni nell’ambito delle tue cure mediche, potresti aver sviluppato anche le seguenti complicanze:3,4,10
- Sovraccarico di ferro, ovvero elevate quantità di ferro nell’organismo, che può causare danni al cuore, al fegato e ad altri organi nel corso del tempo
- Reazioni alla trasfusione, come la febbre
In tutto il mondo le persone convivono con la beta-talassemia

Quasi 290.000 persone in tutto il mondo sono affette da beta-talassemia.10 In passato, la malattia era molto diffusa nei Paesi del Mediterraneo, nel Sud-est asiatico e in Medio Oriente.11 Oggi, soprattutto per le migrazioni delle popolazioni e i matrimoni tra gruppi etnici diversi, la beta-talassemia è presente anche in altre parti del mondo, tra cui il Nord Europa Settentrionale e il Nord America.1,11
In Italia, si contano circa 7.000 persone con la forma grave di beta-talassemia e circa tre milioni di portatori sani. La Sardegna, la Sicilia, le regioni meridionali, ma anche alcune zone del Nord, sono le aree a maggiore diffusione.12
Talvolta, convivere con una malattia complessa, come la beta-talassemia, può farti sentire solo/a, ma ricorda che il personale medico è sempre a disposizione per supportarti, quindi non lo sei. In questo sito web, condividiamo le storie di altre persone che convivono con la beta-talassemia: i loro racconti possono aiutarti a comprendere meglio come vivere la tua vita con la malattia.
